Trajectory inference from simulated data
Robrecht Cannoodt
2016-01-22
Source:vignettes/simulated-data.Rmd
simulated-data.RmdIn this vignette, SCORPIUS is used to infer a trajectory through cells in artificial single-cell RNA-seq data. Note that the dataset is generated in a very naive manner and is only meant to be used for demonstration purposes, not for evaluating trajectory inference methods.
Simulate expression data
Expression values for 384 cells and 500 genes is generated as follows.
library(SCORPIUS)
dataset <- generate_dataset(num_genes = 500, num_samples = 384, num_groups = 4)The resulting dataset is a list containing a matrix named
expression and a data frame named
sample_info.
expression is a 384-by-500 matrix containing the
expression values of all the cells and all the genes.
dataset$expression[1:6, 1:6]## Gene1 Gene2 Gene3 Gene4 Gene5 Gene6
## Sample1 4.229293 0.000000 6.752726 2.911327 9.116873 7.968772
## Sample2 12.063623 6.101313 0.000000 8.023840 6.612786 0.000000
## Sample3 7.973086 12.969934 10.412038 5.564873 10.258806 0.000000
## Sample4 0.000000 10.665796 7.329762 4.635637 9.743435 5.462945
## Sample5 8.467850 12.182375 5.609669 0.000000 7.102293 7.985970
## Sample6 9.269898 9.630904 10.455783 0.000000 8.349845 13.157807sample_info is a data frame with the metadata of the
cells, containing only the group each cell belongs to.
head(dataset$sample_info)## group_name
## Sample1 Group 1
## Sample2 Group 1
## Sample3 Group 1
## Sample4 Group 1
## Sample5 Group 1
## Sample6 Group 1In order to infer a trajectory through this data, SCORPIUS first reduces the dimensionality of the dataset.
Reduce dimensionality of the dataset
SCORPIUS uses Torgerson multi-dimensional scaling to reduce the dataset to three dimensions. This technique attempts to place the cells in a space such that the distance between any two points in that space approximates the original distance between the two cells as well as possible.
The distance between any two samples is defined as their correlation
distance, namely 1 - (cor(x, y)+1)/2. The reduced space is
constructed as follows:
expression <- dataset$expression
group_name <- dataset$sample_info$group_name
space <- reduce_dimensionality(expression, "spearman", ndim = 3)The new space is a 384-by-3 matrix, and can be visualised with or without colouring of the different cell types.
draw_trajectory_plot(space, progression_group = group_name, contour = TRUE)## Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`.
## ℹ See also `vignette("ggplot2-in-packages")` for more information.
## ℹ The deprecated feature was likely used in the SCORPIUS package.
## Please report the issue at <https://github.com/rcannood/SCORPIUS/issues>.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.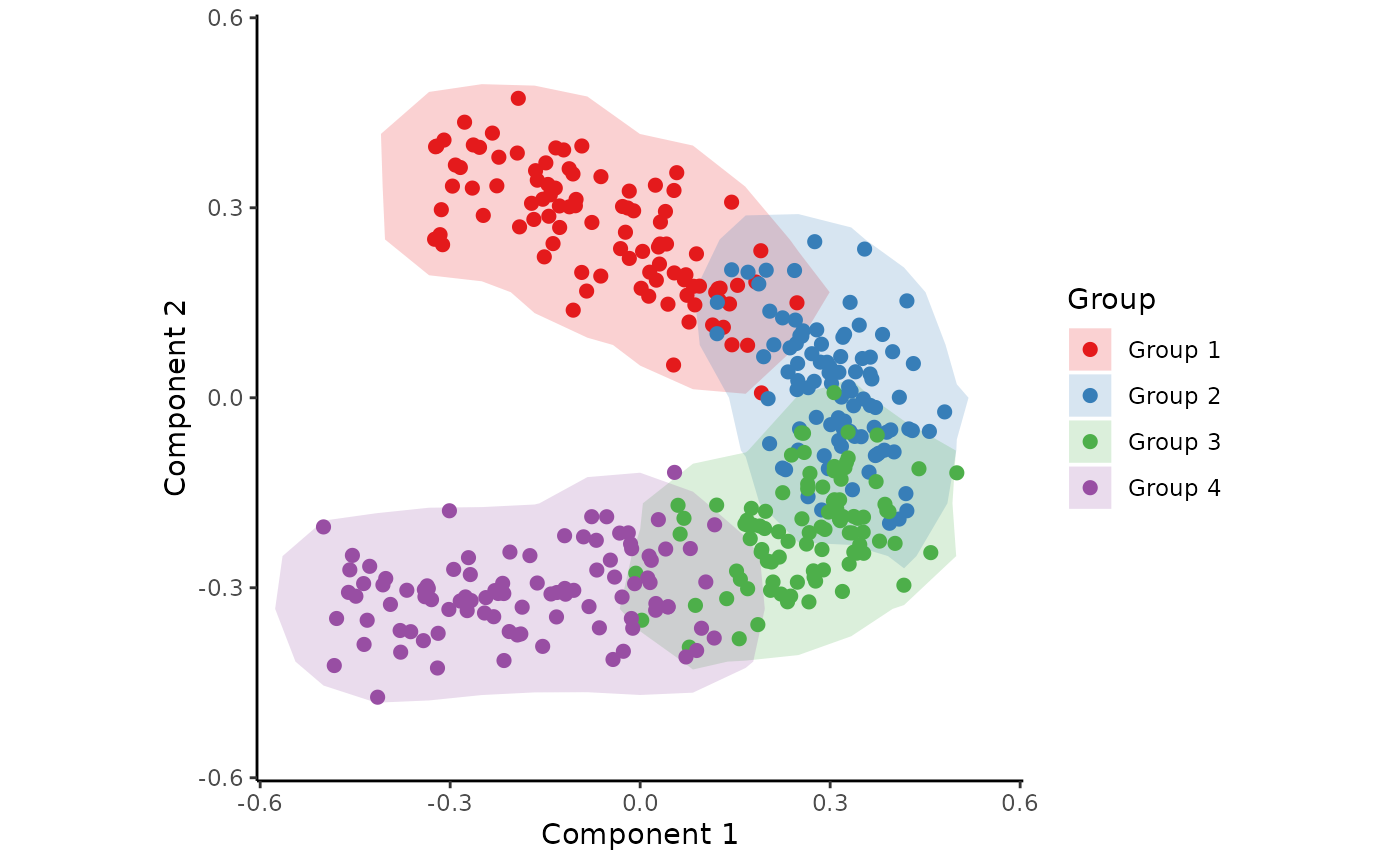
Inferring a trajectory through the cells
The main goal of SCORPIUS is to infer a trajectory through the cells, and orden the cells according to the inferred timeline.
SCORPIUS infers a trajectory through several intermediate steps, which are all executed as follows:
traj <- infer_trajectory(space)The result is a list containing the final trajectory
path and the inferred timeline for each sample
time.
The trajectory can be visualised with respect to the samples by
passing it to draw_trajectory_plot:
draw_trajectory_plot(
space,
progression_group = group_name,
path = traj$path,
contour = TRUE
)## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
## ℹ Please use `linewidth` instead.
## ℹ The deprecated feature was likely used in the SCORPIUS package.
## Please report the issue at <https://github.com/rcannood/SCORPIUS/issues>.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.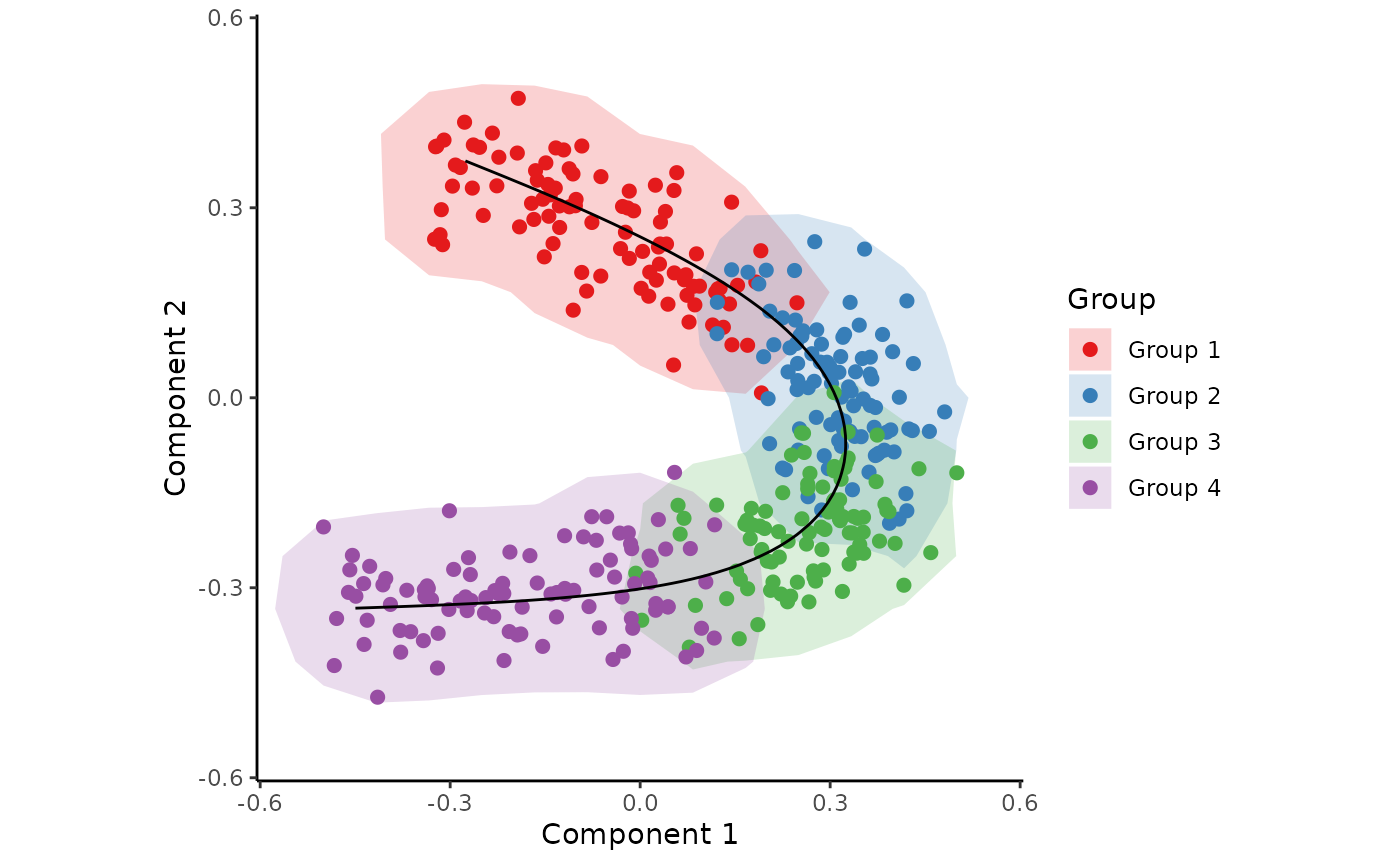
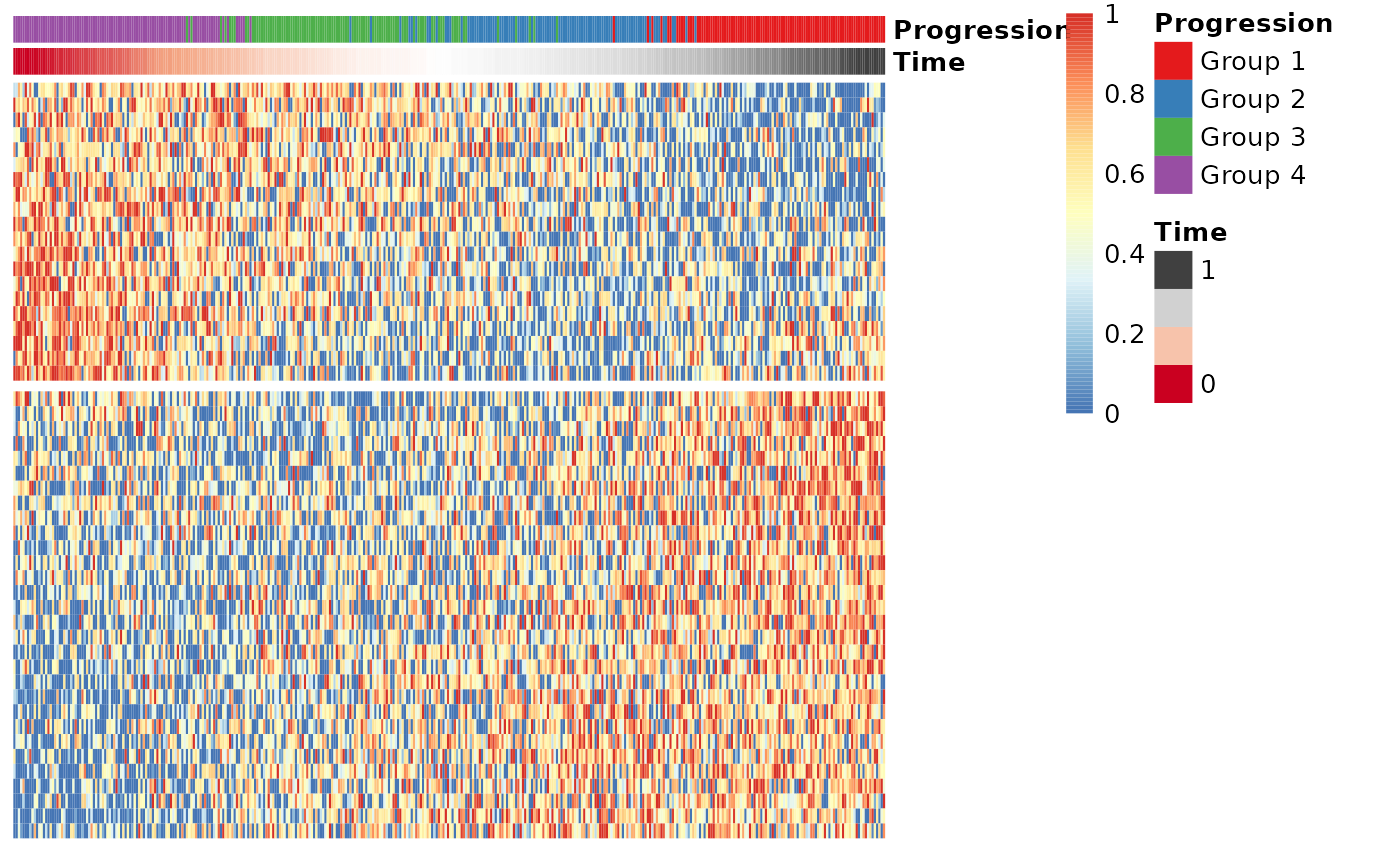
Finding candidate marker genes
We search for genes whose expression is seems to be a function of the trajectory timeline that was inferred, as such genes might be good candidate marker genes for the dynamic process that is being investigated.
gimp <- gene_importances(expression, traj$time, num_permutations = 0, num_threads = 8)
gene_sel <- gimp[1:50,]
expr_sel <- expression[,gene_sel$gene]To visualise the expression of the selected genes, use the
draw_trajectory_heatmap function.
draw_trajectory_heatmap(expr_sel, traj$time, group_name)
Finally, these genes can also be grouped into modules as follows:
modules <- extract_modules(scale_quantile(expr_sel), traj$time, verbose = FALSE)
draw_trajectory_heatmap(expr_sel, traj$time, group_name, modules)